Hemoproteins:
Heme as prosthetic group
The heme group serves to reversibly bind oxygen
- Haemoglobin – RBCs
- Myoglobin – Muscles
- Cytochromes – heme group serves as e– carrier
- Catalase – it catalyses H2O2
Hemoglobin
- Conjugated protein
- Red blood pigment found in erythrocytes
Heme:
- Non protein part
- Prosthetic group
- Contain Protoporphyrin IX with iron at its centre
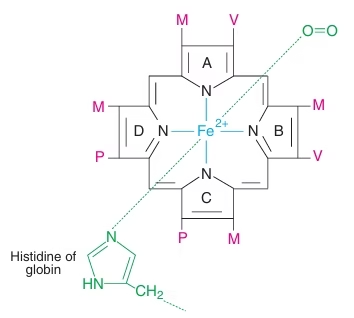 Fig: Structure of Heme
Fig: Structure of Heme
Globin:
- Apoprotein part
- Two types of chains
- 2α –each chain contain 141 AA
- 2β – each chain contain 146 AA
- Total-574 AA
Structure of Hemoglobin:
- Conjugated globular Tetrameric allosteric protein
- Non protein component: Heme; Protein component: Globin
- Four subunits of Hb are held together by non-covalent interactions: hydrophobic, ionic, hydrogen bonds.
- Each subunit contains a heme group.
Functions of hemoglobin:
- Transport of 02 from lungs to tissues
- Transport CO2 and protons from the tissues to the lungs
Types of Normal Hemoglobin
Myoglobin:
- Monomeric oxygen binding hemoprotein found in heart and skeletal muscle.
- Single polypeptide chain with heme moiety.
- Structurally resembles the individual subunits of hemoglobin molecule.
- Functions as a reservoir for oxygen and oxygen carrier that promotes the transport of oxygen to the rapidly respiring muscle
Binding of O2 to hemoglobin:
- One molecule of hemoglobin (with four hemes) can bind with four molecules of 02.
- Each heme moiety can bind with one 02.
- O2 binding is cooperative
O2 dissociation curve (ODC):
- The binding ability of Hb with O2 at different partial pressures of oxygen (pO2) can be measured by a graphic representation known as ODC.
- Ability of Hb to unload & load O2 at physiological pO2 is shown by ODC
- Sigmoid shape of ODC is due to allosteric effect or Cooperativity.
- Binding of oxygen to one heme increases the binding of oxygen to other hemes – Homotropic interaction. This is called Positive Cooperativity.
- Thus the affinity of Hb for the last O2 is about 100 times greater than binding of first O2 to Hb.
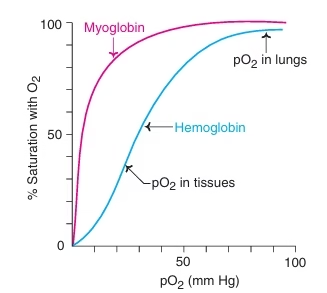 Fig: Oxygen Dissociation Curve of Hemoglobin
Fig: Oxygen Dissociation Curve of Hemoglobin
Quaternary structure of Hb exists in two alternate conformations:
- T state (tense) – with low affinity for O2 – deoxy state
- R state (relaxed) – with higher affinity for O2 – oxy state
- In T subunits – the binding sites are closed.
- In R state – the binding sites are open.
- With successive addition of O2, T shifts the equilibrium towards R state.
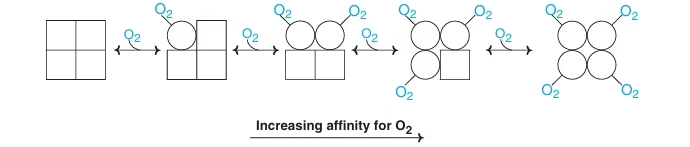 Fig: Cooperative binding of O2 to Hb
Fig: Cooperative binding of O2 to Hb
Factors affecting Oxygen Dissociation Curve
Small molecules that influence the O2 binding capacity of Hb are called effectors/modulators.
Negative effectors: 2,3-BPG, CO2, H+, Cl-
Positive effectors: O2
Right shift (easy oxygen delivery/ O2 affinity is decreased)
- High 2,3-BPG
- High H+
- High CO2
- High Temperature
- HbS
Left shift (give up oxygen less readily/ O2 affinity is increased)
- Low 2,3-BPG
- Low H+
- Low CO2
- Low Temperature
- HbF
The Bohr Effect:
- The binding of oxygen to hemoglobin decreases with increasing H+ concentration (lower pH) or when the hemoglobin is exposed to increased partial pressure of CO2 (pCO2).
- Hb-O2 + H+ → Hb-H+ + O2
- Influence of pH and pCO2 to facilitate oxygenation of Hb in lungs and deoxygenation at the tissues is known as Bohr Effect.
- It is due to a change in the binding affinity of oxygen to hemoglobin.
- Bohr effect causes a shift in the oxygen dissociation curve to the
2,3-Bisphosphoglycerate:
- Negative allosteric effector
- Binds to the central pocket of deoxyHb by interacting with positively charged AA of β- chain.
- Stabilizes deoxygenated Hb by cross linking β- chains.
- Normally present in RBCs and shift the O2 dissociation curve to the right
- Favors the O2 dissociation and its supply to tissues.
Clinical significance of 2,3-BPG:
- In Hypoxia (high altitude, COPD), Anemia:
2,3 BPG levels are elevated, so increased supply of O2 to tissues.
- In blood transfusion:
Stored blood in acid citrate-dextrose: Decreased 2,3 BPG: Fail O2 supply to the tissues.
Addition of Inosine prevents decrease of 2,3 BPG.
- HbF:
- Binding of 2,3 BPG weak: Higher affinity for O2.This facilitates transplacental transfer of O2.
Hemoglobin derivatives
Hemoglobin (specifically heme) combines with different ligands and forms hemoglobin derivatives.
- OxyHb
- DeoxyHb
- Methemoglohin (metHb)
- Carboxyhemoglobin (COHb)
Methemoglobin (metHb):
- Hb (Fe2+) → MetHb (Fe3+)
- Caused by H2O2, free radicals and drugs
- The Methemoglobin (with Fe3+) is unable to bind to 02.
- The occasional oxidation of hemoglobin is corrected by the enzyme methemoglobin reductase present in erythrocyte
Carboxyhemoglobin (COHb):
- CO is a toxic compound that can bind with Hb and has about 200 times more affinity than O2
- Clinical toxicity exhibits when conc. of the carboxyHb exceeds 20%
- Symptoms- headache, nausea, vomiting, breathlessness, irritability etc.
- Treatment– O2 administration
Hemoglobin abnormalities:
- Quantitative abnormalities:
Decrease production of normal hemoglobins
Ex. alpha Thalassemia, beta Thalassemia
- Qualitative abnormalities:
Production of abnormal hemoglobin e.g. sickle cell anemia.
Sickle-cell anemia
- (HbS): two normal α-globin chains and two abnormal (point missense mutation) β-globin chains.
- This is due to a difference in a single amino acid.
- In HbS, glutamate (Polar) at sixth position of β-chain is replaced by valine (Non-polar).
- Glu β6 → Val
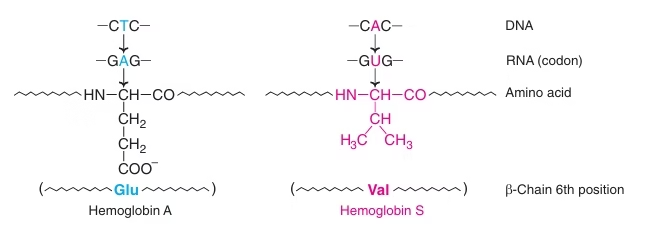 Fig: Molecular basis of Hbs
Fig: Molecular basis of Hbs
- Decrease in solubility of HbS in deoxygenated form
- Homozygous- caused by inheritance of two mutant genes (one from each parent) that code for β-chains.
- Heterozygous- only one gene (of β-chain) is affected while the other is normal.
- The erythrocytes of heterozygous contain both HbS and HbA and the disease is referred to as sickle cell trait.
- The individuals of sickle-cell trait lead a normal life, and do not usually show clinical symptoms.
 Fig: RBCs (A) From a normal person; (B) From a patient with sickle cell anemia.
Fig: RBCs (A) From a normal person; (B) From a patient with sickle cell anemia.
Mechanism of Sickling In Sickle Cell Anemia:
Fig: Mechanism of Sickling In Sickle Cell Anemia
Sticky patch of one deoxyHbS binds with complementary site of another deoxy HbS leading to polymerization of deoxy HbS to form gelatinous network of long fibrous polymer– Distort shape of RBC – sickle shape.
Differences between Sickle cell anemia and Sickle cell trait:
| Sickle cell anemia | Sickle cell trait |
| Individual has received 2 mutant genes, one from each parent | Individual has received 1 mutant gene from parent and normal gene from other parent |
| Homozygous | Heterozygous (AS) |
| Hbs only | HbA + HbS in equal amounts |
| Clinical symptoms
|
No clinical symptoms
At high altitudes – symptoms |
Hb S gives protection against Malaria:
This malarial parasite spends a part of its life cycle in RBCs.
Factors which interrupt the parasites cycle:
- Increased lysis of sickled cells – shorter RBC life span.
- Parasites cause acidity of RBC – increase sickling
- K+ is low in sickled cell – parasite cannot survive
Sickle-cell trait (heterozygous with 40% HbS) provides resistance to malaria – Adaptation for the survival of the individuals in malaria infested regions
Diagnosis:
- Sickling test:
Microscopic examination of blood smear prepared by adding reducing agents such as sodium dithionite.
- Electrophoresis:
- When subjected to electrophoresis in alkaline medium (pH 8.6) , sickle-cell hemoglobin (HbS) moves slowly towards anode (positive electrode) than does adult hemoglobin (HbA).
- The slow mobility of HbS is due to less negative charge, caused by the absence of glutamate residues that carry negative charge.
- In case of sickle-cell trait, the fast moving HbA and slow moving HbS are observed.
Treatment:
- Administration of sodium cyanate inhibits sickling of erythrocyte. Cyanate increases the affinity of 02 to HbS and lowers the formation of deoxyHbS.
- In patients with severe anemia, repeated blood transfusion is required.
Other Abnormal Hb:
- HbC disease
- HbD disease
- HbE disease
Thalassemia:
- Thalassemia are a group of hereditary hemolytic disorders characterized by impairment/ imbalance in the synthesis of globin chains of Hb
- Defect in the production of α-or β-globin chain.
- There is however, no abnormality in the amino acids of the individual chain
Alpha –Thalassemia:
- Caused by a decreased synthesis or total absence of α-globin chain of Hb.
- There are four copies of α-globin gene, two on each one of the chromosome 16.
- Four types of α–Thalassemia occur which depend on the number of missing α-globin genes
Types of alpha- Thalassemia:
| Types | Number of missing genes | Schematic presentation | Clinical symptoms |
| Normal | Nil | -α1 – α2 –
-α1 – α2 – |
Nil |
| Silent carrier | 1 | -α1 – α2 –
-α1 – α2 – |
No symptoms |
| α- thalassemia trait | 2 | -α1 – α2 –
-α1 – α2– |
Mild anemia |
| Hb H disease | 3 | -α1 – α2 –
-α1– α2 – |
Mild to moderate anemia |
| Hydrops fetalis | 4 | -α1– α2 –
-α1– α2 – |
Fetal death |
β– Thalassemia:
- Decreased synthesis or total lack of the formation of β-globin chain causes β-thalassemia.
- The production of α-globin chain continues to be normal, leading to the formation of a globin tetramer (α4) that precipitate. This causes premature death of erythrocytes.
2 types-
- β-thalassemia major
- β-thalassemia minor
β-thalassemia major:
- Homozygous state with a defect in both the genes responsible for β-globin synthesis.
- Healthy at birth since β – globin is not synthesize during the fetal development.
- They become severely anemic and die within 1-2 years.
- Frequent blood transfusion is required for these children.
β-thalassemia minor:
- Heterozygous state with a defect in only one of the two β-globin gene pairs on chromosome 11.
- Also known as β -thalassemia trait
- Usually asymptomatic since the individuals can make some amount of β -globin from the unaffected gene.
Biosynthesis of Heme:
- Erythrocyte producing cells of bone marrow (Erythroid cells): 85%
- Liver: 15 %
- Partially mitochondra /cytoplasm /mitochondra.
Starts with the condensation of glycine and succinyl coenzyme A under the action of a rate limiting enzyme δ-amino levulinic acid synthase. d-ALA will be formed.
Pyridoxal phosphate (vitamin B6) is a coenzyme for this reaction.
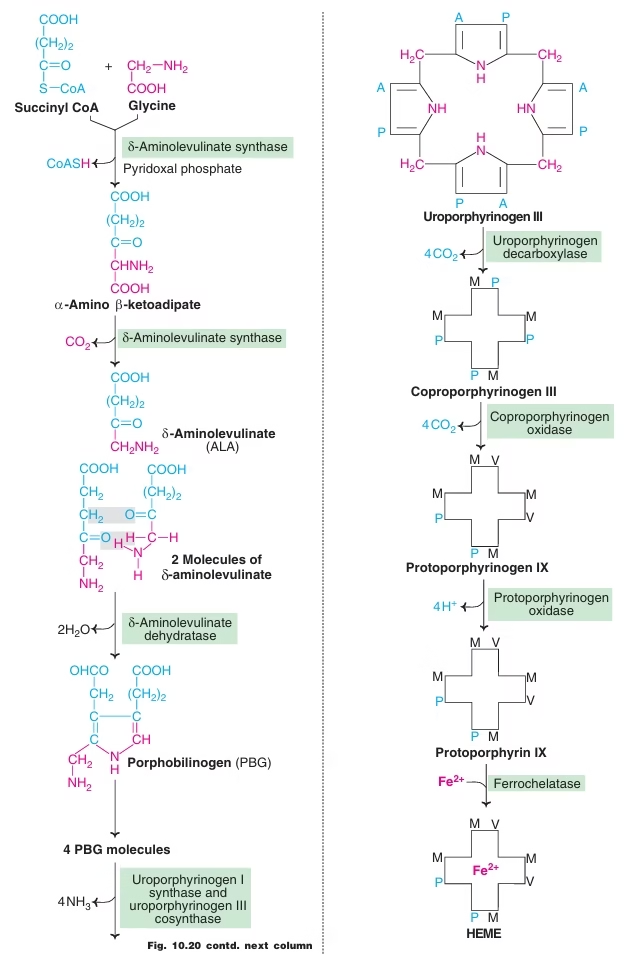 Fig: Heme synthesis
Fig: Heme synthesis
Regulation of Heme Synthesis:
Liver for formation of Hemoproteins (Cyto. P450) whereas Erythroid cells for formation of Hb.
Regulation in Liver:
- Feedback inhibition
- Repression of ALA synthase.
- Inhibition of transport of ALA synthase
Erythroid Cells:
- Uroporphyrinogen synthase
- Ferrochelatase
- Stimulated by cellular uptake of iron.
Porphyria:
Definition: Porphyrias (either inherited or acquired) are the metabolic disorders of heme synthesis, characterized by the increased excretion of porphyrins or porphyrin precursors.
Classified into two categories:
- Erythropoietic: Enzyme deficiency in erythrocytes.
- Hepatic: Enzyme defect lies in the liver.
All porphyrias are Autosomal dominant; Except Congenital erythropoietic Porphyria.
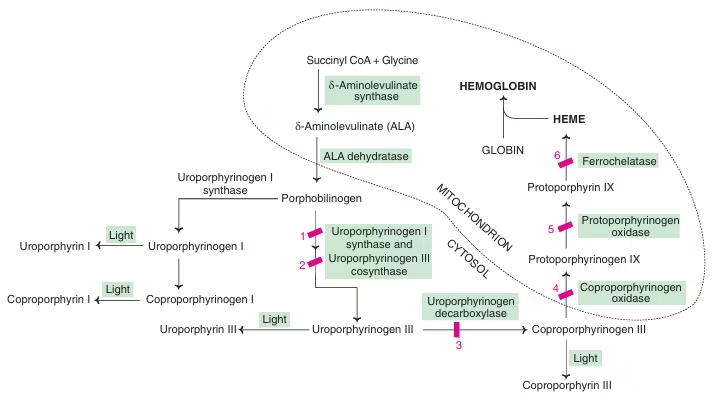 Fig: Heme synthesis and Porphyria
Fig: Heme synthesis and Porphyria
| Hepatic types of porphyria | Enzyme Defect | Clinical features |
| Acute intermittent porphyria | Uroporphyrinogen I synthase | Abdominal pain, neuropsychiatric
symptoms |
| Porphyria cutanea tarda | Uroporphyrinogen decarboxylase | Photosensitivity
|
| Hereditary coproporphyria | Corpoporphyrinogen oxidase | Abdominal pain, photosensitivity,
neuropsychiatric symptoms |
| Variegate porphyria | Protoporphyrinogen oxidase | Abdominal pain, photosensitivity,
neuropsychiatric symptoms |
| Erythropoietic Types of Porphyria | Enzyme Defect | Clinical features |
| Congenital erythropoietic porphyria | Uroporphyrinogen III cosynthase | Photosensitivity, increased hemolysis |
| Protoporphyria | Ferrochelatase | Photosensitivity |
Acute Intermittent Porphyria:
- Hepatic, autosomal dominant
- Caused by a deficiency in porphobilinogen deaminase, which is involved in the conversion of porphobilinogen (PBG) to uroporphyrinogen III
- PBG, uroprophryin, and 5-ALA accumulate in the plasma and the urine
- Patients have neuropyschiatric symptoms and abdominal pain (neurovisceral)
Porphyria cutanea tarda:
- Most common porphyria
- Hepatic, autosomal dominant
- Disease is caused by a deficiency in uroporphyrinogen decarboxylase, which is involved in the conversion of uroporphyrinogen III to coproporphyrinogen III
- Uroporphyrinogen accumulates in urine
- Patients are photosensitive (cutaneous photosensitivity). Accumulation of porphyrinogens results in their conversion to porphyrins by light.
Heme catabolism
Normal red cell destruction:
- Red cell destruction usually occurs after a mean life span of 120 days.
- The cells are removed extravascularly by macrophages of the reticuloendothelial system (RES), especially in the bone marrow but also in the liver and spleen.
The breakdown of red cells liberates:
- Iron for recirculation via plasma transferrin to marrow erythroblasts
- Protoporphyrin which is broken down to bilirubin.
- Globins which are converted to amino acids.
About 6 g of Hb is broken down & resynthesized per day in an adult man.
Fate of Globin:
May be reutilized for Hb formation or broken down to individual amino acids.
Sources of Heme:
The major bulk of heme (about 80%) is released by the breakdown of hemoglobin from the senescent RBCs. The remaining 20% of heme comes from the degradation of myoglobin & cytochromes.
One gram of Hb on destruction finally yields 35 mg of bilirubin. Approximately 250-350 mg of bilirubin is daily produced in adults.
Degradation of heme to Bile pigments:
Transport of bilirubin:
Albumin has two binding sites for bilirubin- a high affinity site & a low affinity site. Approximately 25 mg bilirubin can bind can tightly bind to albumin (at high affinity sites) per 100 ml of plasma.
Albumin-bilirubin complex enters the liver, bilirubin dissociates & taken up by sinusoidal surface of hepatocytes of liver by carrier mediated active transport.
Conjugation of bilirubin:
In the liver, by enzyme bilirubin glucoronyl transferase.
Secretion of bilirubin into bile & Excretion of bilirubin:
Conjugated bilirubin is excreted into bile canaliculi against a concentration gradient which then enters the bile. The transport of bilirubin diglucoronide is an active, energy dependant & rate-limiting process. Almost all the bilirubin that enters bile (>98%) is conjugated.
Fig: Bilirubin Metabolism (Overview)
Jaundice
Disorders of Hemoglobin Catabolism:
Disease or conditions that interfere with bilirubin metabolism may cause a rise in its serum concentration of bilirubin.
Hyperbilirubinaemia:
- When serum bilirubin exceeds 1mg/dl – Hyperbilirubinaemia
- >2.2 to 5mg/dl – Jaundice
Jaundice:
- It is a clinical condition characterized by yellow color of sclera and skin.
- Caused due to deposition of bilirubin due to elevated levels in serum
- Normal range-
Total bilirubin – 0.2 to 1.0 mg/dl
Direct (conjugated) – 0.2 to 0.4 mg/dl
Indirect (unconjugated) – 0.2 to 0.6 mg/dl
Types of jaundice:
- Hemolytic / Prehepatic jaundice
- Associated with increased hemolysis of erythrocytes
- This results in the over-production of bilirubin beyond the ability of the Iiver to conjugate and excrete the same
- increase formation of urobilinogen and stercobilinogen
- Increased indirect bilirubin
- Increased urine urobilinogen
- Dark brown color of feces due to high content of stercobilinogen
- Excess Hemolysis may be due to Incompatible Blood Transfusion, Malaria, Sickle Cell Disease, Deficiency of G-6PD enzyme etc
- Hepatic jaundice:
- Caused by dysfunction of liver due to damage to liver parenchymal cells
- Increased levels of conjugated and unconjugated bilirubin in the serum.
- Dark colored urine due to the excessive excretion of bilirubin and urobilinogen
- SGOT & SGPT Increased
- Causes – Viral hepatitis, cirrhosis of liver, cardiac failure, poisons and toxins, Drugs etc.
- Obstructive / Posthepatic jaundice:
- Due to an obstruction in the bile duct that prevents the passage of bile into the intestine
- Due to the blockage in bile duct, the conjugated bilirubin from the liver enters the circulation
- Increased conc. of direct bilirubin
- Dark colored urine
- Clay colored stool due to absence of stercobilinogen
- Caused by gallstones, Ca head of pancreas, Ca of common bile duct etc
Biochemical Changes in Jaundice:
| Test Name | Hemolytic | Obstructive | Hepatic |
| Serum Bilirubin | Unconjugated ↑ | Conjugated ↑ | Both ↑ |
| Van den Berghs Test | Indirect Positive | Direct positive | Biphasic |
| Bile pigments in urine | Urobilinogen | Conjugated bilirubin | Urobilinogen and conjugated bilirubin |
| Serum Enzymes | ALT, AST, ALP — | ALP↑↑,
ALT, AST marginal ↑ |
ALT, AST ↑↑
ALP marginal ↑ |
Neonatal physiological jaundice:
- Transient condition is the most common cause of unconjugated hyperbilirubinemia
- It results from accelerated hemolysis around the time of birth and an immature hepatic system for the uptake, conjugation, and secretion of bilirubin.
- Not only is the bilirubin-UGT activity reduced, but there probably is reduced synthesis of the substrate for that enzyme, UDP-glucuronic acid.
- Since the increased amount of bilirubin is unconjugated, it is capable of penetrating the blood-brain barrier when its concentration in plasma exceeds that which can be tightly bound by albumin (20–25 mg/dL).
- This can result in a hyperbilirubinemic toxic encephalopathy, or kernicterus, which can cause mental retardation.
- Treatment- Phenobarbitone
- Phototherapy with blue light (420-470nm)
Inherited conditions
Indirect hyperbilirubinemia
- 1. Crigler-Najjar types I and II
- 2. Gilbert’s syndrome
Direct hyperbilirubinemia
- 1. Dubin-Johnson syndrome
- 2. Rotor’s syndrome
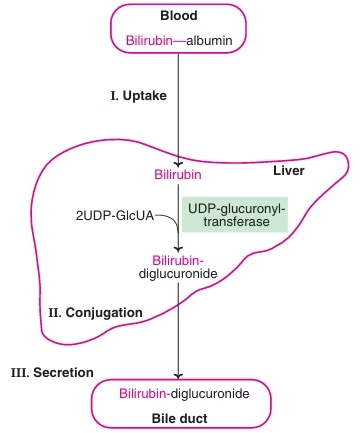 Fig: Diseases of bilirubin metabolism
Fig: Diseases of bilirubin metabolism
Crigler-Najjar syndrome:
- Deficiency of UDP glucoronyl transferase
- Type I & II
- Type I is Fatal
- Unconjugated bilirubin increases more than 20mg/dl
- Children die because of Kernicterus
- Type II is milder form
Gilbert’s disease:
- Impaired hepatic uptake of bilirubin
- Partial conjugation defect
- Reduced activity of UDP glucoronyl transferase
- Asymptomatic and mild
- Unconjugated bilirubin increases – 3mg/dl
Dubin-Johnson syndrome
- Defective excretion of conjugated bilirubin
- Elevation of conjugated bilirubin
- Mutation in the MRP-2 protein responsible for transport of conjugated bilirubin into bile
- Black liver jaundice
Rotor syndrome:
- Similar to Dubin-Johnson syndrome
- Defective excretion of conjugated bilirubin
- Elevation of conjugated bilirubin
- Exact defect is not identified
- No staining of liver